Sabemos que a cranioestenose (craniossinostose) é uma doença que ocorre devido a fusão prematura em uma ou mais suturas cranianas e que ocorre mais comumente como um defeito isolado. Nestes casos, temos as cranioestenoses chamadas de simples: escafocefalia, trigonocefalia, plagiocefalia etc. Se você quer saber sobre as cranioestenoses simples acesse aqui.
Temos ainda as cranioestenoses complexas, onde mais de uma sutura craniana pode estar envolvida (para saber mais sobre cranioestenoses complexas clique aqui). Neste contexto se inserem ainda as craniossinostoses sindrômicas, que além de envolver várias suturas cranianas, estão relacionadas ainda a um conjunto maior de anomalias no corpo em decorrência da própria síndrome genética. Entenda agora sobre estes tipos de cranioestenose e as principais diferenças e preocupações em relação ao seu tratamento, sobretudo quando comparamos com as cranioestenoses simples.
O que são cranioestenoses sindrômicas?
Cranioestenoses complexas são todas aquelas que envolvem o fechamento simultâneo de mais de uma sutura no crânio. São, como o próprio nome já diz, mais complexas e difíceis de tratar. Estas cranioestenoses podem aparecer como formas isoladas ou estarem relacionadas a síndromes genéticas.
Algumas alterações genéticas já sabidamente levam ao fechamento de várias suturas do crânio e adicionalmente estão associadas a outras alterações no corpo (como malformações cardíacas, nos dedos da mão e pé, etc). Quando estas alterações genéticas estão presentes em conjunto com o fechamento das suturas do crânio dizemos que estas são as cranioestenoses sindrômicas, que estão, portanto, inseridas como uma das alterações morfológicas dentro de uma síndrome genética muito maior. Nem todas as cranioestenoses complexas são sindrômicas, porém praticamente todas as cranioestenoses sindrômicas são complexas.
Quais as síndromes genéticas estão mais relacionadas a cranioestenoses?
As síndromes genéticas mais frequentemente associadas à craniossinostose incluem Apert, Crouzon, Pfeiffer, Carpenter e Saethre-Chotzen.
Estas cranioestenoses são frequentemente esporádicas, surgindo como resultado de mutações autossômicas dominantes envolvendo receptores do fator de crescimento de fibroblastos (FGFRs) e genes TWIST. Isso significa que não há necessariamente história familiar desta doença e ela pode ocorrer em famílias que nunca tiveram sequer um caso de cranioestenose.
Abaixo falarei um pouco de cada uma destas principais síndromes. Depois discutiremos os principais problemas e riscos ao sistema nervoso que estas cranioestenoses apresentam, bem como o tratamento cirúrgico necessário.
SÍNDROME DE APERT
A síndrome de Apert (acrocefalosindactilia tipo I) é uma doença autossômica dominante que ocorre em 6 a 15,5 em 1 milhão de nascidos vivos. A maioria dos casos é esporádica. Mutações no gene que codifica o receptor 2 do fator de crescimento de fibroblastos (FGFR2), localizado no cromossomo 10, são responsáveis por quase todos os casos conhecidos. A prevalência da síndrome de Apert aumenta com o aumento da idade paterna.
A cranioestenose relacionada a síndrome de Apert é caracterizada pela fusão prematura de ambas as suturas coronais, resultando em uma forma de crânio braquicefálica. Em geral estes pacientes têm a testa “achatada” e o crânio é mais curto. Além disso, os pacientes podem apresentar graus variados de: exorbitismo (olhos “saltados”), hipertelorismo (olhos afastados) e hipoplasia da face média (o meio do rosto fica “recuado”). As orelhas são de implantação baixa costumam ser acompanhadas por uma estrutura nasal anormalmente pequena e plana com uma ponta bulbosa.
Os pacientes com síndrome de Apert têm ainda sindactilia complexa simétrica de ambas as mãos e pés (dedos “grudados”). Malformações cardíacas, genito-urinárias podem ocorrer, bem como podem ter hidrocefalia em cerca de 10% dos casos. Outras malformações no sistema nervoso central podem existir, tais como agenesia de corpo caloso. Seu quociente de inteligência (QI) varia entre 59 e 94. Deficiência intelectual (= QI <84) e retardo mental (QI <71) são mais frequentemente vistos em crianças com síndrome de Apert em comparação com as demais cranioestenoses sindrômicas e complexas.
SÍNDROME DE CROUZON
A síndrome de Crouzon (disostose craniofacial tipo I) é autossômica dominante e ocorre com uma frequência de 16 em 1 milhão de nascidos vivos. É causada por uma mutação genética no receptor 2 do fator de crescimento de fibroblastos (FGFR2), embora mutações no receptor 3 do fator de crescimento de fibroblastos (FGFR3) tenham sido identificadas em indivíduos com síndrome de Crouzon também. Uma ligação entre a idade paterna avançada e casos esporádicos de síndrome de Crouzon também foi demonstrada, como é frequentemente visto em doenças autossômicas dominantes causadas por novas mutações.
A maioria dos pacientes com Crouzon tem fechamento da sutura coronal bilateral, mas diferentes suturas cranianas podem estar envolvidas, às vezes resultando em pansinostose (fechamento de todas as suturas cranianas). É importante chamar atenção que os pacientes com Crouzon podem desenvolver esta pansinostose após o parto, o que significa que eles têm uma forma craniana normal no nascimento e primeiros meses de vida, o que pode atrasar o diagnóstico. Além disso, os pacientes podem ter também proptose (“olhos saltados”) e hipoplasia da face média (quando o meio do rosto fica “recuado”), mas o grau de deformidade facial é mais leve que o da síndrome de Apert. Também em contraste com a síndrome de Apert, os pacientes com síndrome de Crouzon geralmente têm mãos e pés estruturalmente normais, bem como frequentemente tem a inteligência normal. As anormalidades da coluna cervical ocorrem em até um terço dos pacientes e devem ser cuidadosamente avaliadas antes da correção cirúrgica da craniossinostose.
SÍNDROME DE PFEIFFER
A síndrome de Pfeiffer (acrocefalosindactilia tipo V) é o resultado de um defeito genético autossômico dominante, mais comumente associada a mutações no receptor 1 do fator de crescimento de fibroblastos (FGFR1) e no receptor 2 do fator de crescimento de fibroblastos (FGFR2). Vários pacientes demonstraram uma mutação pontual no locus Ser351Cys (serina alterada para cisteína no códon 351 do aminoácido) do FGFR2, que parece resultar em um fenótipo mais grave com prognóstico reduzido de sobrevida.
As características clínicas da síndrome de Pfeiffer incluem graus variáveis de craniossinostose e hipoplasia da face média (“retração” do meio da face). Polegares largos são frequentemente associados com sindactilia (dedos “grudados”) parcial do segundo e terceiro dedos, assim como do segundo, terceiro e quarto dedos. A presença e extensão da sindactilia são variáveis. Anormalidades esqueléticas (por exemplo, sinostose radio-umeral do cotovelo), no sistema nervoso central (por exemplo, hidrocefalia) e gastrointestinais (por exemplo, ânus imperfurado) também ocorrem.
A síndrome de Pfeiffer é classificada em três subtipos. A classificação é útil para prever o prognóstico de deficiência mental e sobrevivência. Pacientes com síndrome de Pfeiffer tipo 1 têm o fenótipo clássico com craniossinostose bicoronal simétrica, sindactilia variável, polegares largos e dedões dos pés alargados. A inteligência é normal ou quase normal, e a maioria dos pacientes sobrevive até a idade adulta. Os tipos 2 e 3 são mais graves. Além da craniossinostose bicoronal usual, várias outras suturas são frequentemente envolvidas. O tipo 2 especificamente está relacionado ao crânio em trevo, além de outras malformações muito graves nos demais sistemas do corpo. Este tipo está relacionado a prognóstico desfavorável e morte precoce frequente. Em geral Pfeiffer está relacionado a uma deficiência intelectual.
SÍNDROME DE CARPENTER
A síndrome de Carpenter (acrocefalopolissindactilia tipo II), é uma doença autossômica recessiva rara. Está associada a mutações na RAB23 (proteína associada a RAS).
Os pacientes afetados pela síndrome de Carpenter apresentam comumente fechamento das coronais, sagital e lambdoide concomitantes (pansinostose). A ponte nasal é plana e os cantos laterais dos olhos são deslocados lateralmente com ou sem dobras cantais internas. Outras características craniofaciais incluem uma mandíbula e/ou maxila hipoplásica, orelhas de implantação baixa e malformadas e um palato estreito e arqueado alto.
Pacientes com síndrome de Carpenter também são frequentemente afetados pela polidactilia (dedos “extras”). Os achados comuns incluem ainda braquidactilia (dedos curtos) das mãos com clinodactilia (dedos curvos). Anomalias cardiovasculares são comuns, além disso os pacientes tendem a ser obesos e com deficiência mental moderada.
SÍNDROME DE SAETHRE-CHOTZEN
A síndrome de Saethre-Chotzen (acrocefalosindactilia tipo III), é uma doença autossômica dominante. Mutações no gene TWIST foram identificadas em muitos pacientes, bem como mutações no receptor 2 do fator de crescimento de fibroblastos (FGFR2) também foram relatadas.
Os pacientes afetados pela síndrome de Saethre-Chotzen geralmente apresentam craniossinostose das suturas coronal, lambdoide e/ou metópica. A aparência facial característica inclui uma testa elevada (turricefálica), linha do cabelo baixa e assimetria facial. A sindactilia cutânea (dedos “colados”), geralmente parcial, ocorre frequentemente e envolve o segundo e terceiro dedos e/ou terceiro e quarto dedos. A maioria dos pacientes tem inteligência normal, porém alguns casos estão associados a retardo mental geralmente leve.
Que problemas uma criança com cranioestenose sindrômica pode ter?
Pacientes com craniossinostose sindrômica podem ter problemas comportamentais, visuais, e auditivos, além de deficiência mental, conforme já relatado.
A perda auditiva comumente é condutiva causada por otite média recorrente. Costuma ser leve a moderada, sendo encontrada em muitos pacientes com cranioestenoses sindrômicas: 44% em Apert, 28,5% em Crouzon-Pfeiffer, 62,1% em Saethre-Chotzen e 6,7% em pacientes com cranioestenoses complexas não-sindrômicas. Um diagnóstico precoce da perda auditiva é importante por causa de suas possibilidades de tratamento e seu envolvimento no retardo do desenvolvimento da fala e da linguagem.
Os problemas oculares mais frequentes observados em pacientes com craniossinostose são estrabismo e erros de refração. Estrabismo foi detectado em 61,4% dos pacientes e está presente mais comumente em pacientes com Apert. Erros de refração, incluindo erros míopes e hipermetropes, são vistos em 52% dos pacientes com craniossinostose, enquanto o astigmatismo é menos comum.
Pacientes com craniossinostose complexas têm um alto risco de desenvolver hipertensão intracraniana (HIC), possivelmente devido à fusão prematura de múltiplas suturas do crânio associadas ao fechamento de sincondroses da base do crânio levando a uma desproporção entre o tamanho do cérebro e do crânio da criança. Outras causas possíveis são hipertensão venosa devido ao fluxo venoso prejudicado na base do crânio, problemas de circulação ou excesso de líquido cefalorraquidiano (LCR) e apneia obstrutiva do sono (AOS).
A hipertensão intracraniana nestas crianças costuma ser crônica, progredindo ao longo de anos se não for tratada. Ela pode levar a distúrbios no desenvolvimento da cognição e do comportamento. Além disso, a pressão intracraniana aumentada sobre os nervos ópticos pode causar perda visual, levando à cegueira.
Quais os principais objetivos do tratamento cirúrgico de uma cranioestenose complexa?
1. Resolver a desproporção entre o tamanho do cérebro e o crânio – dando mais “espaço” para o cérebro se desenvolver e normalizando a pressão intracraniana.
2. Resolver anormalidades estéticas e funcionais do crânio.
3. Resolver problemas associados a anormalidade craniofacial – tais como: problemas respiratórios, apneia do sono, problemas de oclusão dentária etc.
Como e quando é feita a cirurgia da cranioestenose sindrômica?
Conforme já explicado acima, as cranioestenoses sindrômicas ou complexas não sindrômicas são um grupo muito heterogêneo de doenças. Sendo assim, o tratamento deve ser individualizado para cada paciente, sempre baseado na experiência de um time multidisciplinar, que deve incluir obrigatoriamente um neurocirurgião pediátrico e um cirurgião craniofacial.
De uma forma geral, o tratamento destas condições inclui mais de uma cirurgia (tratamento em múltiplas etapas), realizadas em idades diferentes. Os passos clássicos do tratamento incluem:
- Expansão da parte posterior do crânio
- Tratamento da hidrocefalia (quando presente)
- Avanço frontal ou fronto-orbitário
- Avanço craniofacial
- Procedimentos maxilomandibulares
Nem todas as crianças vão precisar de todas estas etapas, bem como as idades que elas devem ser realizadas variam bastante. Comumente, o primeiro procedimento realizado para estas crianças consiste na expansão da parte posterior do crânio, que costuma ser realizado dentro do primeiro ano de vida. Isto porque neste primeiro momento o neurocirurgião pediátrico está preocupado com a hipertensão intracraniana (HIC). O momento que este procedimento vai ser realizado dentro deste primeiro ano de vida do bebê depende disso. Se houver sinais diretos ou indiretos nos exames clínicos ou de imagem que a pressão intracraniana está aumentada, este procedimento pode ser realizado a partir de 4 meses de vida. Se não houver, ele costuma ser realizado por volta dos 6-8 meses de idade.
Já as cirurgias craniofaciais, que envolvem os avanços da face e da fronte, costumam ser mais tardios, recomendados para crianças a partir de 5 anos, para evitar riscos de novas recorrências e necessidade de mais de uma cirurgia. Entretanto, se a criança apresentar quadros respiratórios obstrutivos graves, proptose severa (olhos saltados para fora) ou algum outro sintoma importante o cirurgião craniofacial pode, em conjunto com o neurocirurgião, decidir antecipar este procedimento.
Como é feito este avanço posterior do crânio – a primeira cirurgia que costuma ser necessária para as crianças com cranioestenoses sindrômicas? Quais são os riscos e as técnicas possíveis?
Existem duas formas básicas para resolver esta situação: a cirurgia clássica com remodelamento craniano e o tratamento com distratores: internos (molas) ou externos.
Na cirurgia clássica o crânio é cortado para liberar a área envolvida com o fechamento das suturas, retirado e remodelado. Depois o neurocirurgião fixa novamente o crânio com placas e parafusos absorvíveis em uma nova posição, dando mais espaço para o cérebro. Esta cirurgia é bem aplicável para crianças menores de 1 ano e não é recomendada para todos os tipos de cranioestenoses complexas, a equipe deve avaliar caso a caso. Ela envolve descolamento do osso em cima de seios durais (veias grossas do crânio) bem delicados, e está relacionada a grande quantidade de sangramento.
Uma alternativa a este procedimento é a colocação de distratores. Nossa equipe tem uma ampla experiência tanto com a técnica clássica quanto com o uso de distratores internos – molas (tecnicamente chamadas de indutores de transformação estrutural – ITE), sendo na nossa opinião, ambas técnicas válidas e muito eficazes, devendo ser analisado risco/benefício para cada criança. A vantagem das molas é que ela pode ser aplicada em qualquer idade e leva a um aumento e remodelamento progressivo do crânio que acompanha o crescimento da criança pelo período que elas são utilizadas.
Quando vamos realizar uma cirurgia com molas inicialmente realizamos um planejamento tridimensional, com a tomografia do paciente, em um software de reconstrução 3D. Conseguimos ver o melhor local de corte e implante destes dispositivos e estimar os efeitos que desejamos atingir no crânio. A cirurgia em si é menos impactante que a técnica clássica, uma vez que o osso não é descolado da dura-máter do paciente, apenas cortado, o que reduz em muito o tempo de cirurgia e o sangramento. Há ainda menor risco de falhas ósseas persistentes e as molas ficam embaixo da pele, sem nada exposto. A desvantagem é que o efeito das molas não é completamente previsível, pois depende das forças que ela e o próprio cérebro irão exercer para remodelar o crânio. Habitualmente as molas são retiradas após um período que varia de 3 a 12 meses.
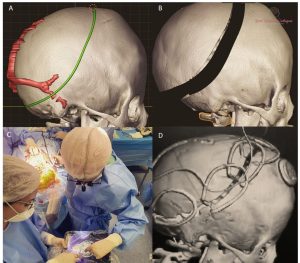
Tratamento de cranioestenose complexa com uso de indutores de transformação estrutural (“molas”). A- Planejamento em software de reconstrução 3d. B- Projeção do resultado final no mesmo software C- Nossa equipe realizando a cirurgia conforme planejamento prévio D- Tomografia de crânio com reconstrução 3d no pós-operatório imediato do paciente mostrando as “molas” bem posicionadas. Fotos de arquivo pessoal – reprodução total ou parcial proibida.
As técnicas podem ainda ser combinadas, utilizando técnicas clássicas de reconstrução com as técnicas dinâmicas, tanto no mesmo, quanto em procedimentos cirúrgicos diferentes.
Como garantir que a pressão intracraniana da criança se normalizou?
Bom, não há como garantir os efeitos a longo prazo da cirurgia na pressão intracraniana, por isso o monitoramento realizado pelo neurocirurgião deve ser contínuo até que o crânio atinja o tamanho adulto (que tenha crescido tudo que devia crescer).
A pressão intracraniana (PIC) pode estar aumentada previamente a cirurgia em 15 a 20% das cranioestenoses simples e até 40% das cranioestenoses complexas. Além disso, existem diversos relatos científicos de cranioestenoses até mesmo simples, que tinham a pressão intracraniana normal na cirurgia, e após anos as crianças apresentaram aumento da PIC. Uma metanálise (estudo de revisão que analisa várias publicações científicas) demonstrou que a PIC pode estar aumentada em cerca de 5% de crianças após cirurgias de cranioestenoses simples (se tiver interesse veja o artigo científico original em inglês clicando aqui).
Por este motivo, é fundamental que o neurocirurgião acompanhe estas crianças, pois garantir a normalidade desta pressão é garantir que o paciente não terá problemas evitáveis no futuro. Existem diversas formas de acompanhar a pressão intracraniana:
1. Exames indiretos – são exames que analisam de forma indireta os efeitos do aumento da PIC. Como exemplos podemos citar: fundoscopia (avaliar papiledema), doppler transcraniano, LCR lombar com manometria, sinais indiretos de aumento da pressão intracraniana em tomografia ou ressonância de crânio (como Chiari, craniolacunias e “prata batida”). A limitação destes exames é que eles podem se alterar apenas em casos mais avançados (graves).
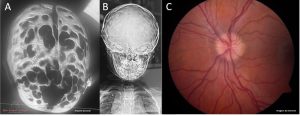
Sinais de aumento da pressão intracraniana. A- Tomografia de crânio com reconstrução 3d mostrando craniolacunias (“buracos” no crânio) B- Radiografia de crânio mostrando sinal da “prata batida” – irregularidades na tábua óssea do crânio C- Fundo de olho com papiledema. Fotos de arquivo pessoal – reprodução total ou parcial proibida.
2. Exames diretos – não existe ainda um exame padronizado e certificado que consiga mensurar diretamente a pressão intracraniana na criança de forma não invasiva. Na verdade, a forma direta de medir a pressão intracraniana é a implantação de um cateter intracraniano no cérebro que seja capaz de medir a pressão. Existem 2 tipos de cateteres:
a. Cateteres temporários – são os mais amplamente disponíveis no mercado. Eles podem ser usados para medir a pressão intracraniana durante a cirurgia, ou podem ficar implantados no crânio da criança por um curto período, ligados a um monitor em uma Unidade de Terapia Intensiva (UTI). Estes cateteres ficam “para fora” da cabeça da criança, por isso há risco de infecção e não podem ficar implantados por longos períodos.
b. Cateteres permanentes – embora não sejam “eternos” são chamados permanentes pois ficam implantados embaixo da pele da criança, podendo funcionar por 2 anos em média. Estes cateteres estão recentemente disponíveis no Brasil, e são conhecidos como cateteres monitores de PIC por telemetria. Eles são uma ferramenta muito válida para crianças com cranioestenoses complexas que a critério do neurocirurgião necessitem monitorização da PIC, podendo ser usados também em outras doenças neurocirúrgicas. Apos a implantação em centro cirúrgico, na consulta do neurocirurgião, no próprio consultório, um leitor é apoiado no cateter e o monitor mostra a PIC e a curva da pressão intracraniana, que pode ser analisada diretamente pelo médico!
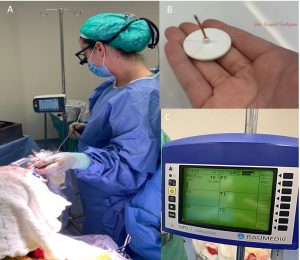
Monitorização da pressão intracraniana em cirurgia de cranioestenose. A- Dra Raquel monitorizando a PIC com cateter temporário durante a cirurgia. B- Cateter de telemetria C- Após cateter de telemetria implantado já conseguimos visualizar a PIC do paciente. Fotos de arquivo pessoal – reprodução total ou parcial proibida.
Se houver suspeita ou comprovação de aumento da PIC, o neurocirurgião pode propor uma nova cirurgia para a criança. Por este motivo, a vigilância do especialista é tão importante.
Quais são os riscos da cirurgia para cranioestenoses sindrômicas?
Os riscos são em geral os mesmos que de uma cirurgia de cranioestenose simples, tais como: perdas sanguíneas importantes (hemorragias) e infecções. Entretanto, vale ressaltar que muitas das crianças com cranioestenoses sindrômicas tem também outras malformações, como por exemplo no coração, rins etc. Estas alterações podem aumentar muito o risco cirúrgico da criança, aumentando as chances de complicações e até de óbito na cirurgia. Por este motivo, antes da cirurgia estas crianças costumam ser avaliadas por médicos de diversas especialidades para definir se a criança tem condições clínicas de ser submetida a cirurgia.
Meu filho foi diagnosticado muito velho com esta doença – existe idade limite para cirurgia?
As cranioestenoses complexas/sindrômicas conforme já foi dito costumam ser tratadas com mais de um procedimento cirúrgico. De modo geral, o primeiro procedimento costuma ser realizado entre 6-8 meses de idade. No entanto, esta idade varia muito de acordo com a condição clínica da criança, experiência da equipe multidisciplinar, comorbidades associadas e número de suturas envolvidas.
Nas cranioestenoses sindrômicas o diagnóstico é mais simples pois em geral a criança tem outras alterações faciais ou corporais que estão ligadas a síndrome genética. Entretanto, em casos mais sutis, se outras malformações não são visíveis, muitas vezes os pais e médicos não especialistas acham o crânio da criança “esquisito” ou “diferente”, mas não conseguem reconhecer que estas alterações são de fato patológicas. É comum crianças mais velhas sem diagnóstico procurarem tardiamente o especialista com sintomas de aumento da pressão intracraniana.
Mesmo em idades mais avançadas existem técnicas cirúrgicas possíveis para aumentar o tamanho do crânio e tratar o aumento da pressão intracraniana, portanto, nunca é tarde para buscar o especialista.
Quando procurar um neurocirurgião?
Sempre procure um neurocirurgião pediátrico o mais breve possível quando houver qualquer alteração no formato da cabeça de seu filho e a suspeita de cranioestenose. Isso permitirá o diagnóstico e tratamento precoces com melhor resultado estético.
 – Agendamento de consulta de primeira vez presencial – ligação em horário comercial nos telefones – (11) 5041-1322ou (11) 5041-9988 / Agendamento automatizado pelo Whatsapp (11) 940208602.
– Agendamento de consulta de primeira vez presencial – ligação em horário comercial nos telefones – (11) 5041-1322ou (11) 5041-9988 / Agendamento automatizado pelo Whatsapp (11) 940208602.